药物偶联物简介
药物偶联物,特别是抗体药物偶联物(ADC),因其临床效果和潜在商业价值而受到广泛关注。而技术的进步导致了药物偶联物新旧理念的交织碰撞,甚至对现有的理念和技术提出了挑战。
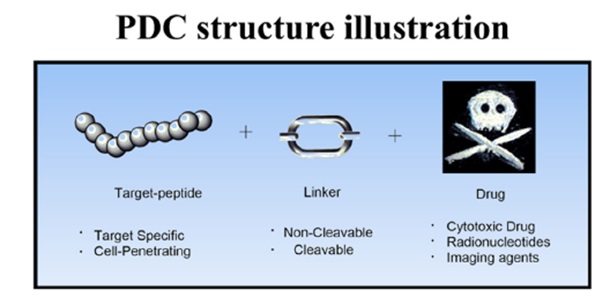
如今,出现了多种新的缀合技术概念,包括肽药物缀合物(PDC)、小分子药物缀合物(SMDC)、免疫刺激抗体缀合物(ISAC)、抗体-寡核苷酸缀合物(AOC)、放射性核素药物缀合物( RDC)、抗体片段-药物偶联物(FDC)、适体药物偶联物(ApDC)、抗体细胞药物偶联物(ACC)、病毒样药物偶联物(VDC)等。此外,新的技术形式如抗体降解剂偶联物( ADeC)仍在不断涌现。本文简要介绍了几类药物偶联物的技术特点和代表性项目开发进展。
抗体药物偶联物 (ADC)
抗体药物偶联物(ADC)是目前最成功的药物偶联物类型,上市药物数量最多,具有良好的临床效益和商业价值。根据2021年《Nature Reviews Drug Discovery》发表的文章,到2026年全球ADC药物市场将达到164亿美元
ADC 旨在通过基于抗体靶向将细胞毒性药物引入癌细胞周围来减少全身暴露并提高安全性。该药物由三个主要成分组成:抗体(靶向)-连接器(将抗体与有效负载连接)-有效负载(杀死肿瘤细胞)。
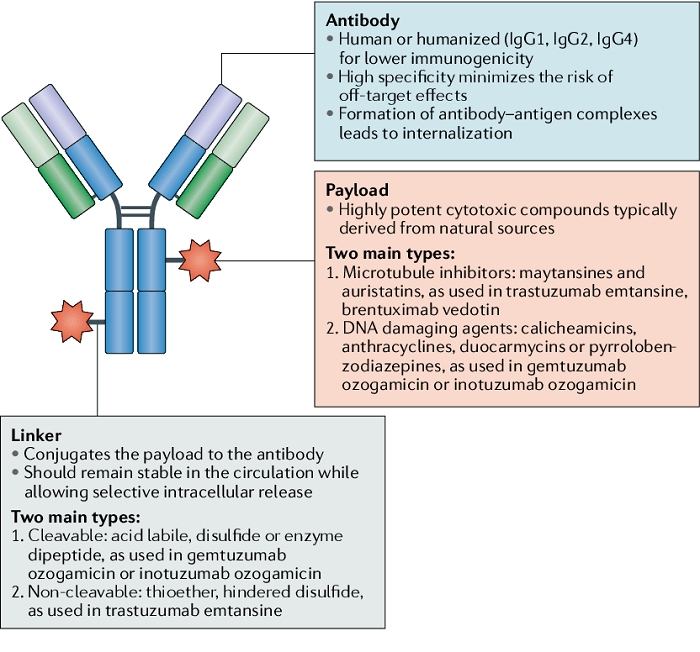
图1|抗体-药物缀合物构建体。图片来源:参考文献1
ADC药物的开发最早,但随着基于更先进技术开发的药物临床验证数据的增加,其面临的挑战也最大。首先,人们普遍认为抗体靶标应该被很好地内吞,但免疫刺激抗体偶联物 (ISAC) 表明也许不需要靶蛋白内吞。其次,传统观点认为抗原必须过度表达,而正常细胞不表达或低表达,而今年 ASCO 会议上 disitamab vedotin 的亚组分析显示,它对几乎所有 HER2 阳性和 HER2 低表达乳腺癌都有益处,以及Enhertu 也适用于 HER2 阳性和 HER2 低表达的肿瘤物种。第三,弹头的品种已经丰富,并不一定需要细胞毒性,免疫刺激剂和调节剂(STING、TLR、Treg)、蛋白水解靶向嵌合体(Protac)、寡核苷酸等药物也在临床或临床前研究中显示出初步有效性。
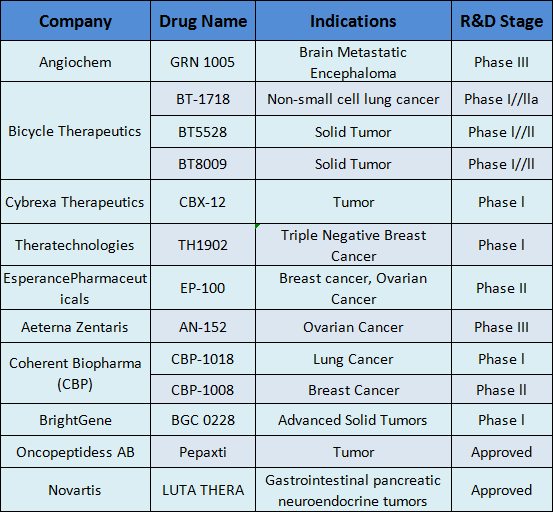
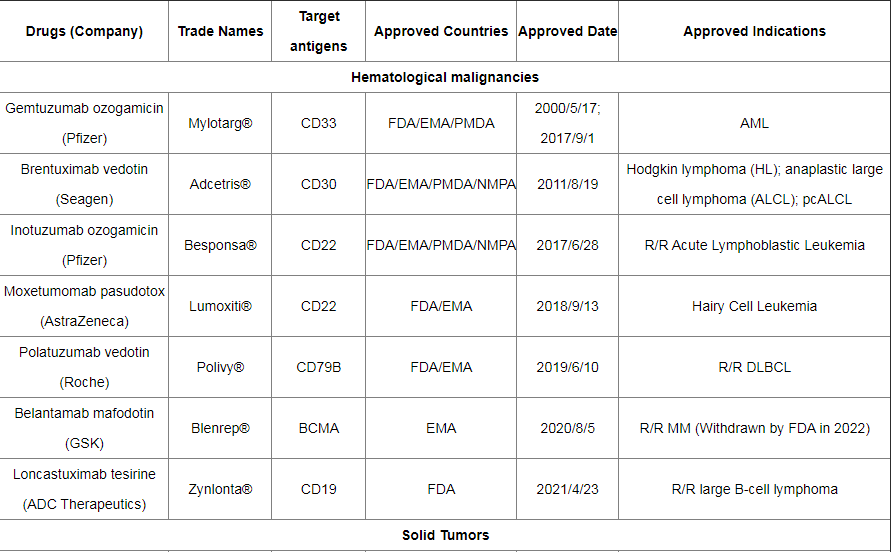
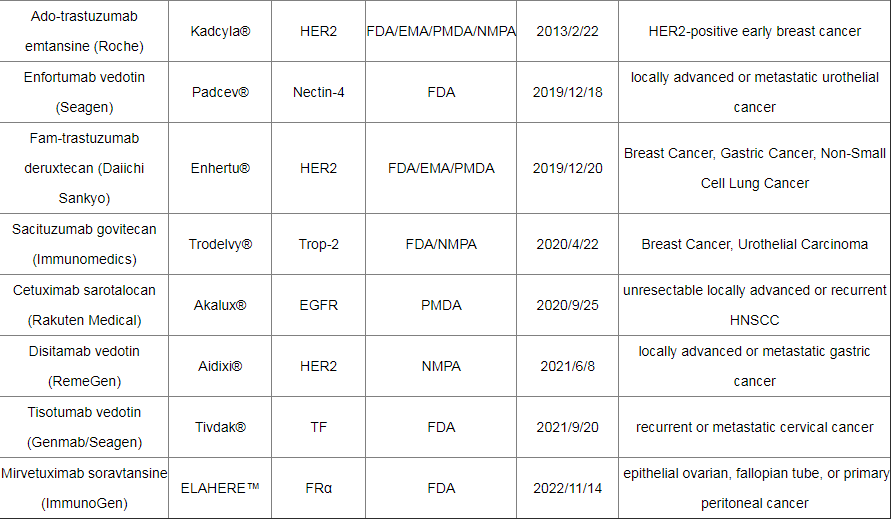
截至2022年12月,全球已有15种ADC药物获批上市,以及超过400种已公布的在研ADC候选药物,主要集中在肿瘤、罕见病和血液学治疗领域。有 136 种候选药物专注于共同靶标,其中 53 种针对HER2。
放射性核素抗体偶联物 (RAC)
放射性核素药物偶联物 (RDC) 与 ADC 类似,它们使用抗体或小分子(包括肽)介导的靶向来精确靶向细胞毒性/成像因子(放射性核素放射性同位素),以避免全身暴露的潜在危险。不同之处在于RDC负载是放射性核素,可用于诊断和治疗功能。其成分也与ADC略有不同,需要添加螯合毒素的特定官能团结构(螯合剂)。一般来说,它仍然由配体-连接体-有效负载组成。
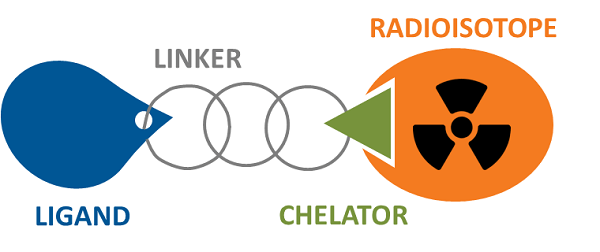
图 2. 放射性核素药物结合物 图片来源:imagingprobes
诺华2017 年以 39 亿美元收购了 Advanced Accelerator applications,其 RDC 药物 Lutathera(镥(177Lu)氧曲肽)自上市以来已成功商业化。 2018 年 10 月,以 21 亿美元收购 Endatory 后,又收购了其 PSMA 靶向放射性配体疗法 177Lu-PSMA-617。
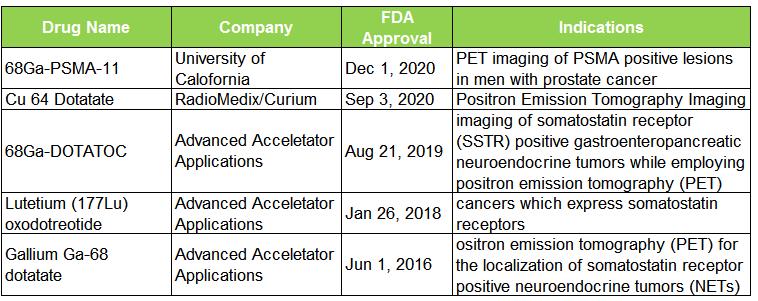
表 1. 近五年 FDA 批准的 RDC
在今年的 ASCO 会议上公布 VISION 研究结果后,177Lu-PSMA-617 被FDA授予突破性疗法称号。在转移性去势抵抗性前列腺癌的治疗中,177Lu-PSMA-617 显着改善了中位影像学无进展生存期(8.7 vs. 3.4 m)并延长了 OS,并将影像学进展或死亡风险降低了 60%。
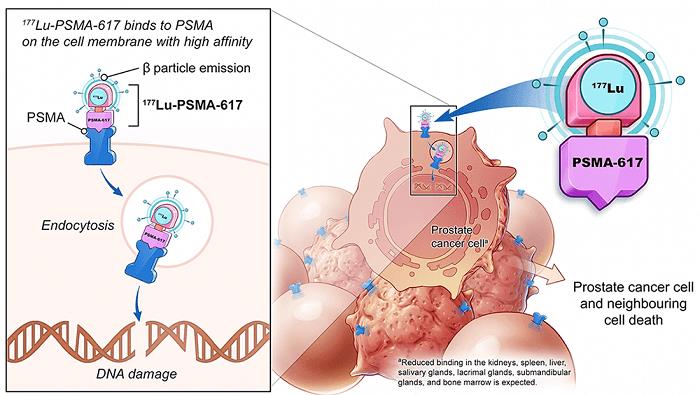
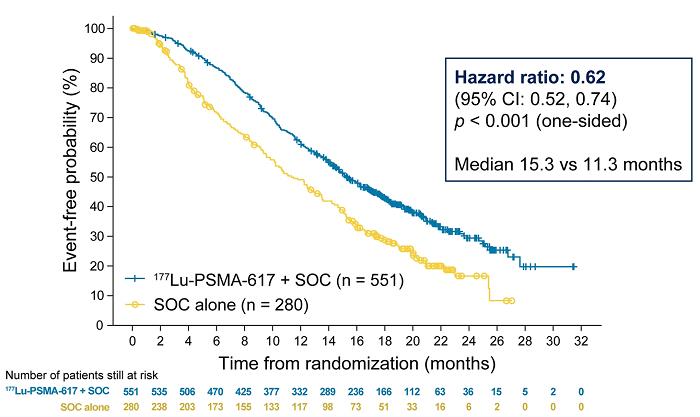
图 3 和图 4。Lutetium-177-PSMA-617,图像来源:ASCO 2021:
小分子药物偶联物 (SMDC)
小分子药物偶联物(SMDC)通常也由靶分子、接头和效应分子(细胞毒性分子、E3连接酶等)组成。
图 5. 小分子-药物缀合物,图片来源:Network of Cancer Research
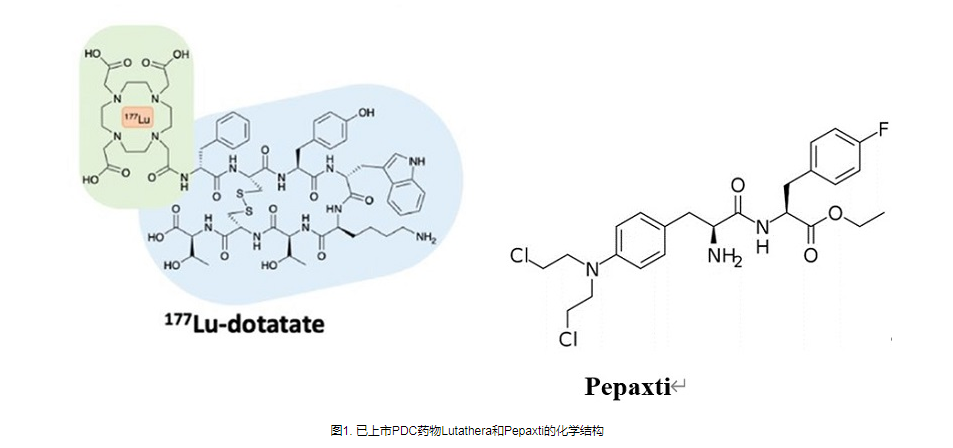
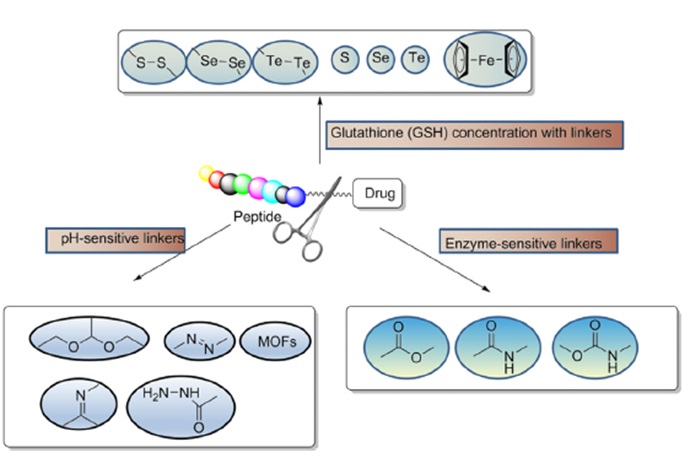
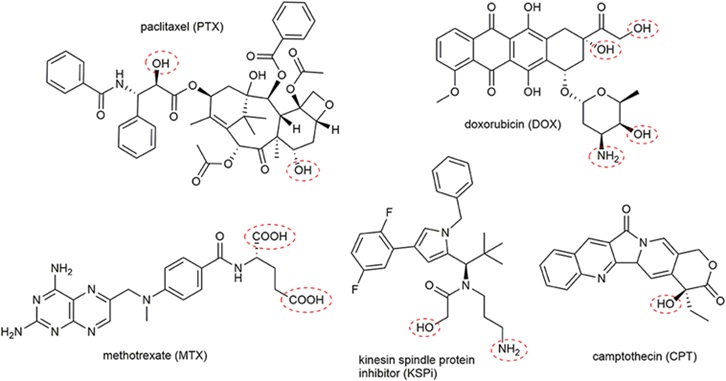
事实上,目前药物偶联物的过度细分也导致了不同概念药物之间的交叉。例如,肽药物偶联物(PDC),通常仍然属于小分子药物偶联物。 Lutathera,177Lu-PSMA-617,虽然根据毒素被归类为RDC,但其靶向配体均属于小分子领域。最近,PEPAXTO获批上市,Oncopeptides将其定位为肽-药物缀合物,但其分子结构并不是药物缀合物组合物的通常形式,或者应该属于前药缀合物(Pro-DC)或前药,在癌细胞周围分解,达到烷化剂样的肿瘤杀伤作用。
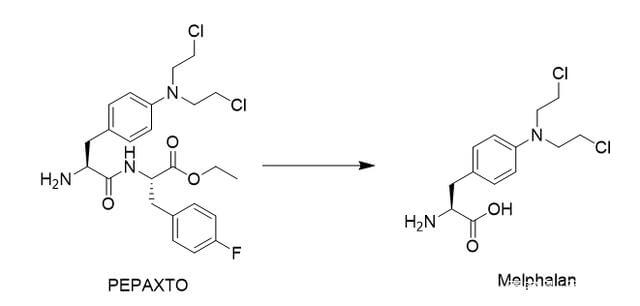
图 6. PEPAXTO 的结构
因此,我们结合了小分子药物偶联物和肽药物偶联物的分析。小分子领域,Endatory产品Vintafolide有条件上市,但临床三期研究失败后撤市。
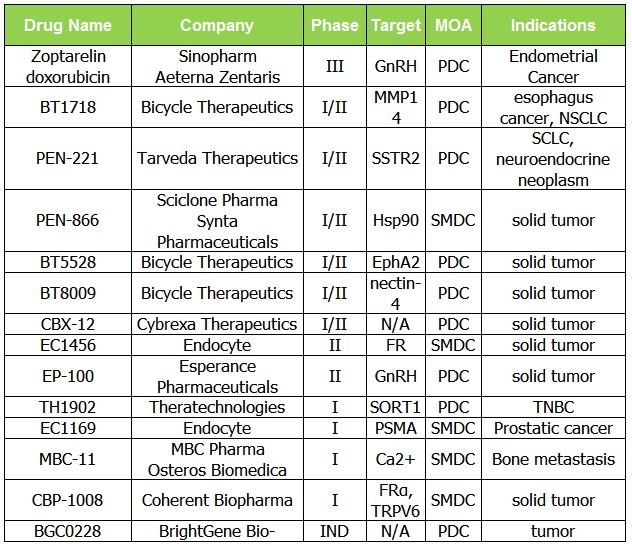
表 2. 正在调查的 SMDC 和 PDC
免疫刺激抗体偶联物 (ISAC)
免疫刺激抗体偶联物(ISAC)的技术要求与ADC非常相似,不同之处在于ISAC负载的是先天免疫激动剂或调节剂,能够将冷肿瘤转化为免疫热肿瘤。此外,其功能与肿瘤微环境激活药物偶联物(TMAC)部分相似,均通过调节免疫刺激和微环境来实现免疫杀伤激活和治疗增敏。
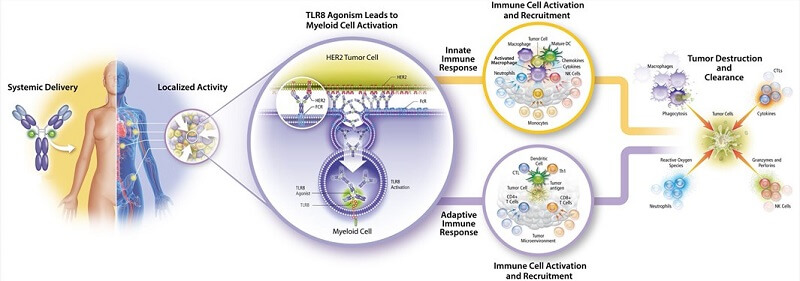
图 7. 免疫刺激抗体偶联物,图片来源:AACR2021
目前参与此类机制的药物主要有Toll样受体激动剂(TLR)型ISAC药物SBT6050、SBT6290、BDC-1001。 STING激动剂ISAC药物XMT-2056、Treg细胞调节ISAC药物ADCT-301等。不过,其中很多药物也被企业自己定义为ADC药物,或许也是因为两者在术语上并没有太多区别药物的外观性能和技术。

图 8. BDC-1001,图片来源:BOLT BIOTHERAPEUTICS
抗体降解剂偶联物 (AdeC)
2021 年 6 月 16 日,瑞士公司 Debiopharm 和韩国公司 Ubix Therapeutics 联合宣布开展研究合作,结合 Multilink 和 Degraducer 两个专有技术平台开发 Antibody Degraducer Conjugates (ADeC)。
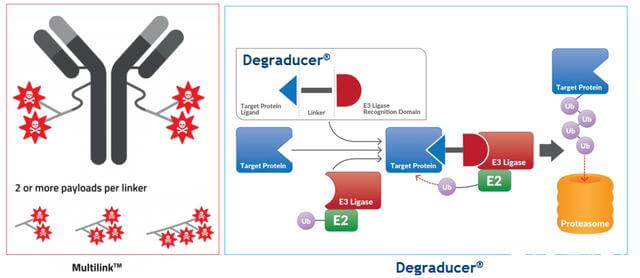
图 9. 抗体-降解剂-缀合物
这种合作才刚刚开始,或许相关药物还没有被研究出来。然而,根据他们的平台技术,预计将开发的ADeC药物将是一种抗体药物缀合物,用降解分子取代有效负载,或许还携带其他有效负载以产生协同效应。 ETC。
ADeC的目的还在于将降解的分子携带至靶位点,避免全身暴露,甚至克服Protac分子的一些潜在成药性问题,如理化缺陷、特异性、PK等。
在 AdeC 领域,Orum Therapeutics 已开始临床前研究,并于最近完成了 8400 万美元的 B 轮融资,以继续推进其产品线。
图 10. Orum 疗法
Orum Therapeutics 的技术还结合了与抗体偶联的蛋白质降解剂,其概念与 Debiopharm 和 Ubix 的技术相似,特别是降解剂,两者都具有泛素酶降解作用机制。
然而,两者也有所不同,Orum Therapeutics 将类别定义为抗体 neoDegrader 缀合物 (AnDC)。
图 11. 抗体-neoDegrader-缀合物,来源:Orum Therapeutics
抗体片段-药物偶联物 (FDC)
抗体片段-药物偶联物(FDC),顾名思义,就是用较小的抗体片段(单链scFv)来替代较大的抗体分子。人们普遍认为抗体片段相对容易找到,并且可以通过生物工程实现更高的 DAR。
图12.抗体片段-药物偶联物(FDC),来源:antikor网站
FDC在技术上与ADC几乎相同,但使用更小的片段抗体有望提高肿瘤渗透性并最大限度地提高药物疗效。小碎片和缺乏 Fc 可以在正常组织和循环中快速清除,从而降低毒性。
适体药物偶联物 (ApDC)
适体药物偶联物 (ApDC) 是药物偶联物的一种形式,它使用结构化寡核苷酸序列作为相应分子的靶标。核酸适体被称为“化学抗体”,具有与抗体类似的靶向和靶点结合特性。与抗体相比,核酸适配体还具有稳定性高、免疫原性低、生产成本低、易于化学修饰等诸多优点。
图 13. 适体药物缀合物 (ApDC),来源:分子疗法:核酸
由于ApDC药物使用寡核苷酸序列,因此它们在接头和缀合策略方面可能与ADC药物不同,但在药物成分、作用机制和有效负载方面与ADC药物没有太大区别。
病毒样药物偶联物 (VDC)
病毒样药物结合物(VDC)是药物结合物的一种形式,它使用设计为非感染性蛋白质纳米颗粒(病毒样颗粒(VLP))的病毒衣壳作为有效的递送载体。
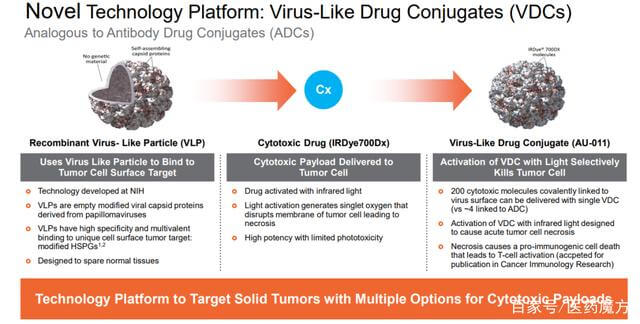
图 14. 病毒样药物偶联物 (VDC),来源:Aura 网站
Aura采用人乳头瘤病毒(HPV)衍生的VLP选择性附着在修饰的硫酸乙酰肝素蛋白聚糖(HSPG)表面,以实现与实体瘤细胞或转移瘤的结合,但不与正常组织结合。AU-001是该机制的VDC产物。病毒样成分选择性地与HSPG结合,结合的红外光激活细胞被激活,选择性地破坏肿瘤细胞,导致肿瘤细胞急性坏死,同时激活免疫系统产生抗肿瘤反应。

图 14. AU-011,来源: Aura 网站
抗体-寡核苷酸缀合物 (AOC)
抗体-寡核苷酸缀合物 (AOC) 是治疗性寡核苷酸利用抗体将siRNA(siRNA、PMO等)递送至特定细胞或组织,从而减少治疗患者疾病所需的药物量,并解决不可靶向和寡核苷酸递送的问题。寡核苷酸与靶向配体的缀合还可以改善寡核苷酸(治疗性RNA或DNA分子)的药代动力学特性并扩大其应用。与ApDC不同的是,AOC旨在实现寡核苷酸的靶向递送,阿斯利康已经对相关产品进行了研究。从技术上来说,AOC使用抗体作为传递介质,也可以假设小分子(包括肽)、蛋白质(酶)等也可以发挥相关功能。细分时,仅以寡核苷酸作为有效负载的药物也产生了各种概念产品。

图 15. 抗体-寡核苷酸缀合物 (AOC),来源:avidity biosciences 网站
Avidity还基于此理念开发了AOC产品AOC1001,用于治疗强直性肌营养不良1型(DM1)疾病,并计划于2021年下半年进行相关临床研究。
结论
总而言之,现阶段的药物缀合物仍然保留相同的组成,即“配体-连接子-效应物”形式。由于技术的进步和药物剂型的繁荣,配体、连接臂和效应分子的定位有了更多的选择,导致了该领域的细分,并出现了多种药物缀合物的表达方式,如ADC、RDC、SMDC、 ISAC、ADeC、PDC、FDC、VDC、AOC 等。但过度的细分也造成了产品概念的混乱,即使是同一产品,不同企业的定义也存在差异。
本质上,大多数药物缀合物都是通过定位配体来达到靶向目的,以及不同功能的效应分子来达到治疗价值或临床目的;产品设计理念延续了ADC药物思路,不同的是三类成分(配体-连接体-效应分子)的转化。不过,ADC、RDC、SMDC和ISAC仍然是最成功的药物偶联物类型,并且已经有相关药物上市或多个临床药物处于概念验证阶段,而其他药物偶联物仍更多处于概念或阶段目前尚处于临床前阶段,能否实现临床价值还有待观察。